
Erik Thiede
Asst. Professor of Chemistry and Chemical Biology, Cornell University
Date and time: December 3rd, 2025 Wednesday at 8am PST / 11am EST / 4pm GMT / 5pm CET / 12am China
For the zoom links, please join the One World Cryo-EM mailing list.
Deconstructing Algorithms for “Discrete” Heterogeneity in Cryo-EM
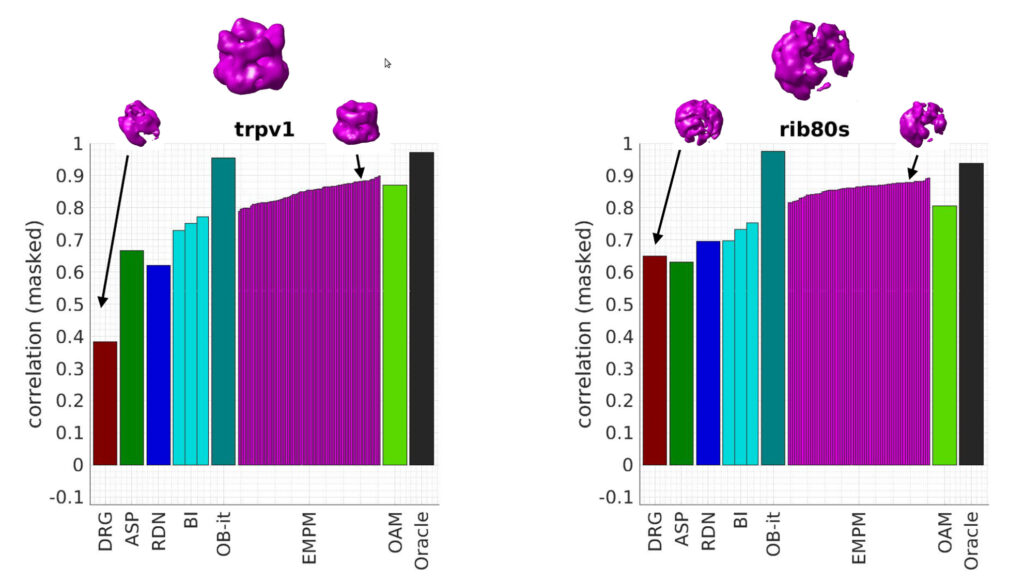
Classic approaches to analyzing conformational or compositional heterogeneity in cryo-EM model attempt to recover a discrete set of structures from the data. But despite how common these algorithms are, exactly how these structures relate to the structures the molecule observes in solution is not fully understood. In this talk, we revisit the likelihood-based algorithm for finding discrete classes. By considering the limit of a large number of structures, we show that this algorithm can be viewed as an approach to solving a stochastic inverse problem for the latent structural probability density. This gives us a new understanding of how the algorithm behaves in the limit of large data. Moreover, our analysis gives a systematic way to construct new algorithms for recovering probability densities associated withheterogeneous data. As an initial contribution, we introduce an algorithm based on the Wasserstein gradient flow. Our work points the way towards more efficient and noise-robust algorithms for recovering heterogeneity with quantitative accuracy.
Diego Sanchez Espinosa, Erik Thiede, Yunan Yang.